Le médicament de A à Z
Aujourd’hui, il existe un arsenal thérapeutique extrêmement riche dans le champ du cancer : chimiothérapie, immunothérapie, thérapies ciblées… Vous vous êtes peut-être déjà demandé quel était le parcours d’un médicament, depuis sa découverte jusqu’à sa mise sur le marché. Comment cela se passe-t-il ? Quelles étapes un médicament doit-il franchir avant d’arriver dans une pharmacie ? Combien tout cela coûte-t-il ?
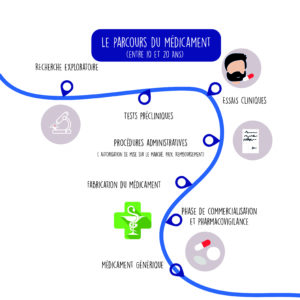
En 2018, le chiffre d’affaires du marché du médicament atteignait 28,9 milliards d’euros1. Dans notre pays, 3,1 milliards2 de boîtes de médicaments sont vendues chaque année, le paracétamol en tête. Si ces chiffres donnent le tournis, ils révèlent avant tout l’importance majeure des médicaments dans notre société. De la découverte d’un candidat-médicament à son arrivée sur le marché, il faut compter environ entre dix et quinze ans. Essais pré-cliniques puis cliniques, autorisation de mise sur le marché, fixation du prix et remboursement, les médicaments passent par de nombreuses étapes avant d’arriver dans nos armoires à pharmacie ou dans celles de l’hôpital.
recherche et essais cliniquesvoir plus
La découverte d’une molécule : recherche fondamentale et recherche appliquée
La recherche fondamentale, qui se fait le plus souvent dans des laboratoires universitaires publics, étudie le fonctionnement normal et pathologique des organismes. La recherche appliquée, privée ou publique, a pour but d’identifier une molécule qui agira sur un mécanisme spécifique et donc potentiellement sur une pathologie précise. Une fois la molécule d’intérêt obtenue, divers procédés pour ce faire existent, cette dernière va suivre un long cheminement pour devenir un médicament commercialisable.
Il est important de garder en tête que sur 10 000 molécules testées, une seule sera commercialisée3.
La phase pré-clinique
Dans l’ordre, il s’agit d’abord de tester la molécule in vitro, sur des cultures cellulaires en laboratoire, puis in vivo, sur l’animal. La phase pré-clinique concerne l’évolution de la molécule dans l’organisme et l’évaluation de sa toxicité.
La phase clinique
Une fois qu’on a évalué la pharmaco-cinétique (le devenir de la substance active dans l’organisme) de la molécule en phase pré-clinique, elle est prête à être testée chez l’homme. On va s’assurer de sa sécurité et de son efficacité chez des volontaires. Cette phase clinique correspond à celle des essais cliniques, ou études cliniques. Pour en savoir plus, nous vous invitons à consulter les fiches « Essais cliniques » de notre site. Ces essais sont aujourd’hui très encadrés par la loi pour garantir la protection des individus y participant. Des comités de protection des personnes (CPP) et les instances règlementaires nationales, qui sont chargés de rendre un avis en amont de toute recherche clinique sur l’homme, relisent attentivement chaque protocole avant sa mise en place. Voici un exemple de procédure de consultation du comité des patients de la Ligue contre le cancer : La phase clinique comprend 4 phases successives :
- Phase 1 : Concernant les thérapies anti-cancéreuses, les molécules sont testées uniquement sur des volontaires malades. Cette phase a pour but d’observer le comportement de la molécule dans l’organisme et d’évaluer sa toxicité (la dose maximale tolérée). Tous les effets secondaires potentiels sont observés.
- Phase 2 : la molécule est expérimentée sur un plus grand nombre de volontaires malades (entre 40 et 100), afin d’évaluer la dose minimale efficace, les effets indésirables et le bénéfice thérapeutique – par bénéfice on entend diminution ou stabilisation de la maladie.
- Phase 3 : la molécule est destinée à être expérimentée sur une population plus importante de volontaires malades (de quelques centaines à quelques milliers). On compare souvent la cohorte de patients traités avec un groupe témoin prenant un traitement de référence ou un placebo. C’est cette étape qui permet de générer les données nécessaires à l’évaluation du rapport bénéfice/risque.
- Phase 4 : c’est une phase de pharmacovigilance (c’est-à-dire la surveillance et l’évaluation des effets secondaires) qui aura lieu après l’autorisation de mise sur le marché. Elle concerne un nombre beaucoup plus large de personnes qui prennent leur traitement dans le cadre de leur prise en charge thérapeutique. Elle observe l’utilisation du médicament sur le long terme dans des conditions réelles d’utilisation : effets indésirables rares, complications tardives…
autorisation de mise sur le marché et prix du médicamentvoir plus
L’autorisation de mise sur le marché (AMM)
Une fois les phases pré-clinique et clinique passées (hormis la phase 4), la structure à l’initiative du médicament dépose un dossier de demande d’autorisation de mise sur le marché (AMM) auprès des autorités compétentes : • au niveau national, c’est l’Agence nationale de sécurité du médicament (ANSM) • au niveau européen, c’est l’Agence européenne du médicament (EMA) Ces instances vont examiner la qualité et l’efficacité de la molécule active et du produit fini. À ce stade, l’ANSM ne se préoccupe pas du prix du médicament.
Cette étude, qui prend généralement environ un an, sert à établir la balance bénéfice/risque qui doit être satisfaisante : en médecine, on applique toujours le principe du « primum non nocere » (d’abord, ne pas nuire).
L’autorisation précoce et l’autorisation compassionnelle
Jusqu’en juillet 2021, l’ANSM pouvait délivrer une autorisation temporaire d’utilisation (ATU) pour un médicament n’ayant pas encore obtenu l’AMM mais qui s’avérait efficace dans des pathologies graves, sans autre alternative de traitement. Les médicaments soumis à une ATU n’étaient pas délivrés en pharmacie de ville. La loi de juillet 2021 a simplifié le système complexe des ATU en distinguant l’autorisation précoce et l’autorisation compassionnelle (on ne parle plus d’ATU).
L’autorisation précoce
L’autorisation précoce concerne les nouvelles molécules présumées innovantes dans une maladie grave, rare ou invalidante pour laquelle il n’existe pas de traitement approprié, ou alors dans une extension d’indication (nouvelle indication pour un médicament existant, c’est-à-dire son utilisation pour une autre pathologie que celle pour laquelle il a été mis sur le marché). Le laboratoire propriétaire de la molécule s’engage à déposer une demande d’AMM dans un délai de deux ans.
L’autorisation compassionnelle
L’autorisation compassionnelle concerne des médicaments (nouvelles molécules ou autres) qui n’ont pas vocation à être commercialisés pour l’indication donnée et qui ne sont pas destinés à obtenir une AMM en France. Cela concerne donc les médicaments habituellement administrés pour une autre pathologie mais dont les effets peuvent être bénéfiques pour une autre.
Les autorisations précoces et compassionnelles permettent à des patients en impasse thérapeutique de bénéficier d’un traitement à titre exceptionnel.
La fixation du prix et le remboursement
Une fois l’AMM obtenue, la Haute Autorité de santé (HAS) intervient, via sa Commission de transparence. Cette dernière, composée de médecins, de pharmaciens, d’épidémiologistes, va évaluer deux éléments : • le niveau de service médical rendu (SMR) • l’amélioration du service médical rendu (ASMR)
Le niveau de service médical rendu (SMR)
Le niveau de service médical rendu répond à la question suivante : le médicament est-il suffisamment intéressant cliniquement pour être remboursé par la collectivité ? Il prend en compte différents critères : efficacité, effets indésirables, pathologie(s) concernée(s), intérêt pour la collectivité… Il existe quatre niveaux de SMR :
- I : important
- II : modéré
- III : faible
- IV : insuffisant
En fonction du niveau de SMR, le taux de remboursement est accordé par l’Union nationale des caisses maladies (UNCAM). Le ministère de la Santé prend ou non la décision d’inscrire le médicament sur la liste des spécialités pharmaceutiques remboursables pour une durée de cinq ans. Plus le niveau de SMR est important, plus le médicament est remboursé par l’Assurance maladie.
L’amélioration du service médical rendu (ASMR)
L’amélioration du service médical rendu évalue ce qu’apporte le médicament par rapport aux thérapies déjà existantes. L’AMSR comporte cinq niveaux :
- Niveau I : amélioration majeure
- Niveau II : amélioration importante
- Niveau III : amélioration modérée
- Niveau IV : amélioration mineure
- Niveau V : amélioration inexistante
Pour les médicaments ayant une ASMR élevée et qui s’avèreront coûteux, la Commission d’évaluation économique et de santé publique (CEESP) de la HAS rend un avis médico-économique qui répond à la question suivante : quel est le rapport entre le coût du médicament et son bénéfice pour la collectivité ? L’ASMR sert de base à la fixation du prix du médicament, qui s’élabore entre le laboratoire pharmaceutique qui a créé le médicament et le Comité économique des produits de santé (CEPS) pour une durée de cinq ans.
Pour résumer, le taux de remboursement et le prix d’un médicament sont fixés en consultation avec des instances indépendantes en fonction de son rapport bénéfice/risque et de son intérêt face à des thérapeutiques déjà existantes.
que se passe-t-il après la mise sur le marché du médicament ?voir plus
La veille sanitaire et le suivi
Une fois le médicament disponible, sa surveillance ne s’arrête pas là. Des études de pharmaco-vigilance seront réalisées par le laboratoire promoteur (celui qui a mis la molécule sur le marché) et des organismes de recherche, notamment en ce qui concerne les nouvelles thérapies. Il s’agit de voir comment le médicament se comporte dans la population générale, hors essais cliniques. Les médecins traitants, les patients et les associations de patients ont leur rôle à jouer pour évaluer les effets à long terme d’un médicament. En cas d’effets indésirables non mentionnés, tous ces acteurs peuvent faire une note auprès du centre régional de pharmacovigilance. On s’intéressera également aux facteurs de risques, aux comorbidités éventuelles, à l’observance du traitement, etc. L’ANSM réévalue régulièrement le médicament après l’AMM en fonction de ces évolutions. Et l’HAS apprécie le service médical rendu (SMR) tous les cinq ans.
En bref, le médicament est scrupuleusement surveillé après sa mise sur le marché.
Brevet et domaine public
Lorsqu’un médicament est découvert, le laboratoire détient un brevet de propriété intellectuelle pour une durée de vingt ans. Avec toutes les longues étapes que nous avons détaillées, le médicament, dans les faits, a un brevet exclusif sur le marché d’environ sept à huit ans. Une fois cette date passée, il tombe dans le domaine public et d’autres laboratoires peuvent fabriquer des génériques, qui sont une copie (non stricte) du médicament princeps. Les génériques comportent les mêmes principes actifs (molécules actives) que le médicament original. Seuls les excipients (composés inactifs qui donnent la texture et le volume, par exemple), les colorants et les conservateurs peuvent changer. Le laboratoire qui fabriquera le générique déposera une demande d’AMM auprès d’une agence du médicament. Le prix du générique est souvent moins cher, car il n’inclut pas les études longues et coûteuses qu’impliquent la découverte et le développement d’une nouvelle molécule.
Apparus en 2006 et différents des génériques car fabriqués à partir d’organisme vivants, les médicaments biosimilaires sont comme leur nom l’indique similaires à un médicament biologique de référence (hormones, vaccins) dont le brevet est tombé dans le domaine public. Leur intérêt est avant tout économique, car ils coûtent moins cher que le traitement de référence et préviennent également les problèmes d’approvisionnement4.
Médicaments anti-cancéreux, où en est-on ?
De plus en plus de traitements anticancéreux sont administrés par voie orale (voir la fiche « J’ai un traitement oral à prendre« ). Cela représente 50 % des traitements anti-cancéreux, seuls ou en association avec des voies injectables par perfusion5. En 2020, plus de 347 000 personnes ont été traitées par chimiothérapie6 par voie orale ou perfusion. L’industrie pharmaceutique investit fortement dans le traitement contre le cancer, avec des innovations majeures (immunothérapie, thérapies ciblées, thérapies géniques) en cours ou à venir.


Catherine, 58 ans, patiente engagée
La prise en charge du cancer (tous types confondus) par l’Assurance maladie représente 16,3 milliards d’euros en 2018 sur 142 milliards d’euros de remboursement général, soit environ 11,5 %7. Du côté de l’industrie pharmaceutique, le marché mondial de l’oncologie est en première place, avec un chiffre d’affaires d’environ 139,4 milliards d’euros en 20208. Ces chiffres sont tous à prévoir à la hausse, d’une part en raison du nombre croissant de malades à soigner, d’autre part en raison du coût des traitements innovants.
Catherine, 58 ans, patiente engagée
POUR PLUS D’INFORMATION, VOUS POUVEZ CONSULTER LES SITES SUIVANTS
|
Le site des entreprises du médicament (LEEM) |
|
Le site de la Haute Autorité de santé (HAS) |
Sources
- https://drees.solidarites-sante.gouv.fr/sites/default/files/2021-03/13-16.pdf
- « Pourquoi la vente de médicaments à l’unité fait autant débat », Les Échos, décembre 2019.
- « 20 questions sur les essais cliniques », LEEM, mai 2020.
- https://www.ameli.fr/assure/sante/medicaments/comprendre-les-differents-medicaments/connaitre-medicaments-biosimilaires
- https://www.abbviepro.com/fr/fr/services-pharmacies/services-pharmacies/pathologies-and-environment/traitements-anticancereux-par-voie-orale/traitements-anticancereux.html
- « Les chiffres clés de la chimiothérapie », Institut national du cancer, 2021.
- https://assurance-maladie.ameli.fr/sites/default/files/2020-07_rapport-propositions-pour-2021_assurance-maladie_1.pdf
- « Pourquoi les laboratoires investissent de plus en plus le marché du cancer », Le Monde, septembre 2021.